DASHR reports the annotation, expression and evidence for specific RNA processing (cleavage specificity scores/entropy) of sncRNA genes, precursor and mature sncRNA products across different human tissues and cell types. Simply input your sncRNA of interest (in a variety of formats) and click Search. By default, a) summary table; b) expression profiles; c) entropy/specific processing information, and d) structure will be produced as outputs for each locus. Users can also view the outputs via an expression browser.
DASHR implementation
The DASHR database was implemented using MySql database engine, while all the scripts were written in PHP (website), AWK and BASH scripting languages. R (v3.3.3) is used for plotting and visualization. DASHR v2.0 is implemented on Amazon Cloud (m3.large instance with Intel Xeon E5-2670 2.5GHz CPUs and 8GB RAM) storing 10TB of raw sequencing data and with processed data stored in MySQL. The database schema can be viewed here.
Methods - Small-RNA-sequencing and annotation pipeline
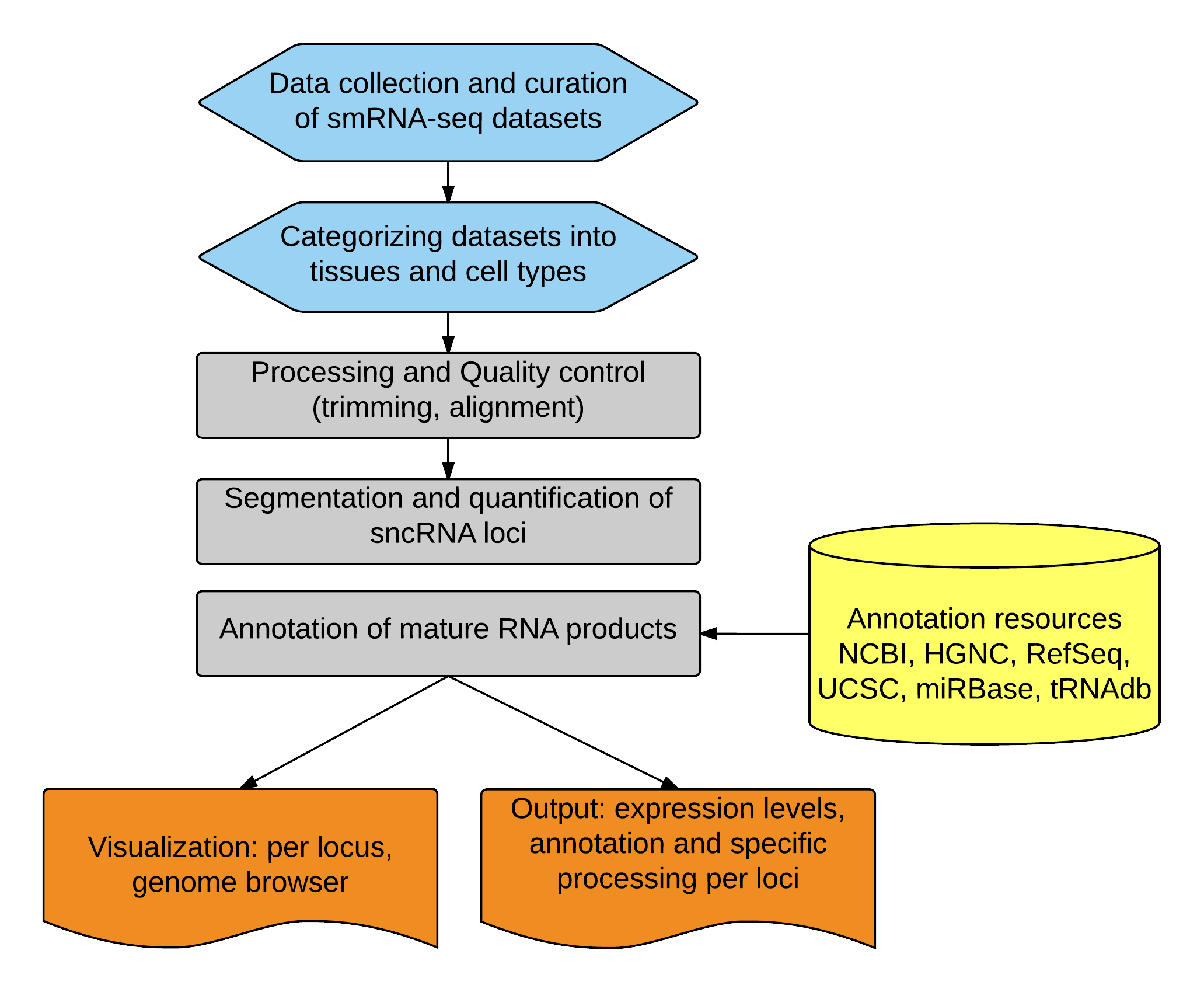
Annotation resources
DASHR integrates multiple existing annotation resources for small non-coding RNAs including miRNA annotations from miRBase (v19); snRNA, snoRNA, scRNA and rRNA annotations are from UCSC genome browser; and GENCODE; tRNA annotations
tRNAdb; and piRNA annotations from NCBI. The current annotation in DASHR also includes tRNA fragment annotations that are created based on the 50nt sequences upstream (5p) and downstream (3p) of the known tRNA genes. Information from NCBI,
HGNC, RefSeq and UCSC was used to build the ID conversion table for each processed and mature sncRNA entry in DASHR.
Data collection and curation of smRNA-seq datasets
We manually curated Illumina smRNA-seq datasets from Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA). These datasets were obtained from non-diseased human tissues and cell types and were generated for studying or profiling small non-coding RNAs. The full list of included samples (smRNA-seq libraries) categorized by tissue and cell type is shown in the Data page.
Categorizing datasets into tissues and cell types
We then manually categorized the smRNA-seq samples into different groups of tissues and cell types, stratified by their experimental sources (i.e. Study ID found in Data summary page).
Processing smRNA-seq datasets
After curation and categorization, we then used SPAR, a modified pipeline built upon CoRAL to process the smRNA-seq datasets and generate sncRNA expression levels. The pipeline can be summarized in three parts: 1) processing and quality control; 2) segmentation and quantification; and 3) annotation.
Processing and Quality control (trimming, alignment)
We first identified the correct adapter (Illumina adapter column found in Data summary page) and trimmed the sequencing reads. We then mapped the trimmed reads to a standardized version of the human reference genome GRCh37/hg19 and GRCh38/hg38. The reads were aligned using STAR allowing for multi-mapping. Over 80% of the trimmed reads were mapped to the human genome on average per dataset (Data summary page details trimming and alignment statistics for each smRNA-seq dataset).
Segmentation and quantification of sncRNA loci
As one of our goals was to identify mature sncRNA products (i.e. peaks with evidence of specific processing patterns, e.g. with low 5p read entropy) and quantify their expression levels in addition to precursor sncRNA gene expression levels, we developed a customized approach to identify peaks with evidence of specific processing for mature sncRNA products in base pair resolution. After identifying the mature sncRNA locations, we then quantified the number of reads falling within these regions as read counts for each sncRNA. To enable comparison across tissues, we took into account the library size information (i.e. number of mapped reads) for each of the sequencing experiments (found in Data summary page) and reported the read count in "reads per million" (RPM).
Annotation of mature RNA products
We overlapped each peak with evidence of specific processing (identified in the previous step) with DASHR annotations (see Annotation resources). Each peak was assigned to its precursor RNA class and sncRNA gene.
Outputs
Summary table
The summary table contains annotation information, including the sequence, genomic coordinates, and structural information for the selected locus. This table also includes summary statistics for expression information, highlighting the total number of experiments with observed expression, total raw read count and total reads count in reads per million (RPM).
Expression profiles
For any set of the specific tissues/cell types, users can view expression profiles or read coverage profiles representing detailed expression information for any sncRNA locus across the set of tissues/cell types. There are also links to the UCSC genome browser, allowing users to view the genomic locus with mapped sequencing data across all tissues.
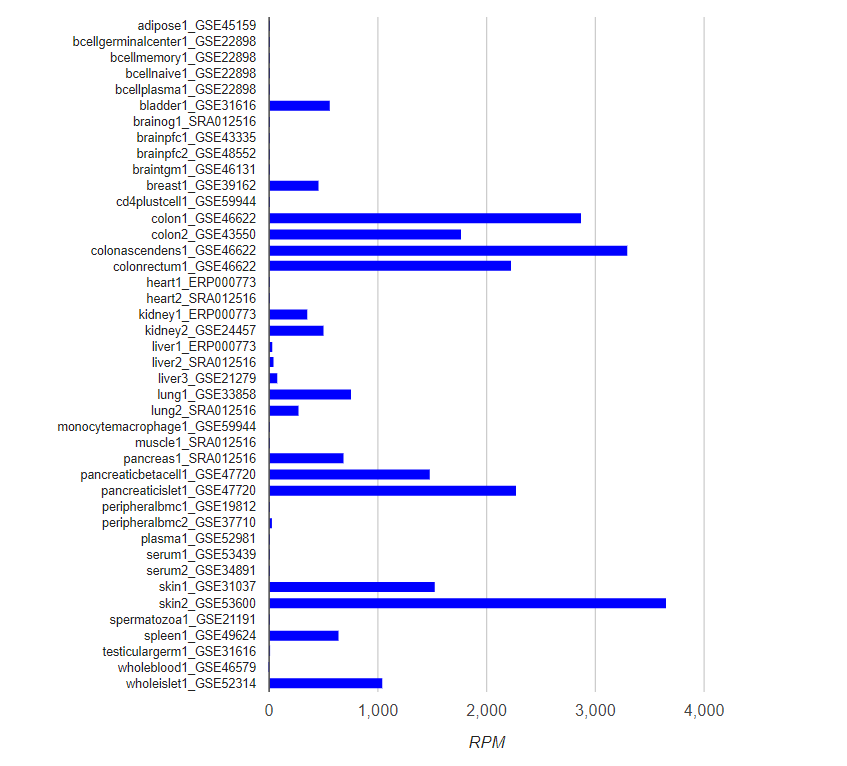
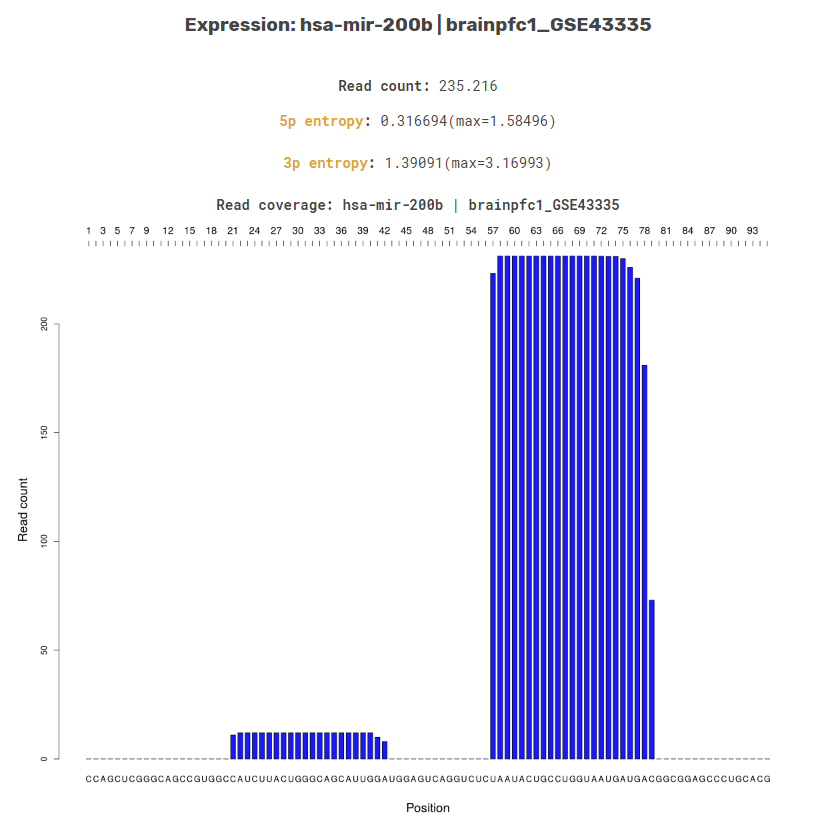
The expression profile plot shows the reads in RPM across all tissues in DASHR for a given locus. Users can choose to display the expression profile in a bar-plot or heatmap format, in which the RPM values are shown.
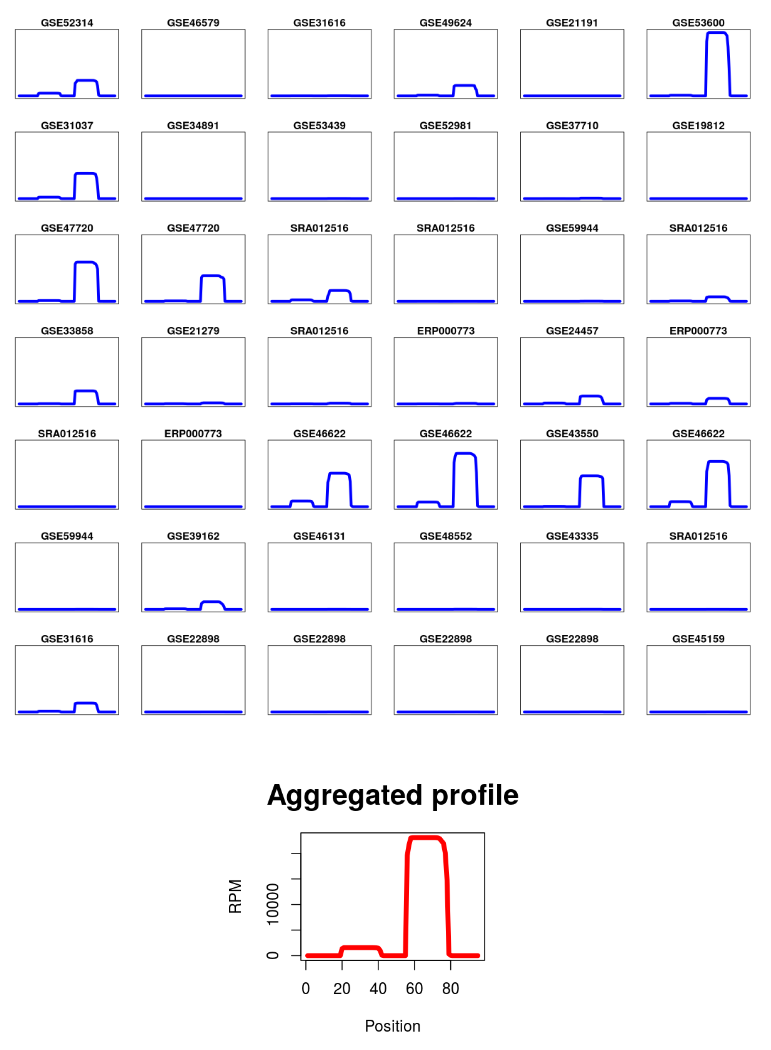
For anyone (or all) chosen specific tissues/cell types, users can view the detailed expression information represented by expression profiles or read coverage profiles.
Expression Profile Across Tissues
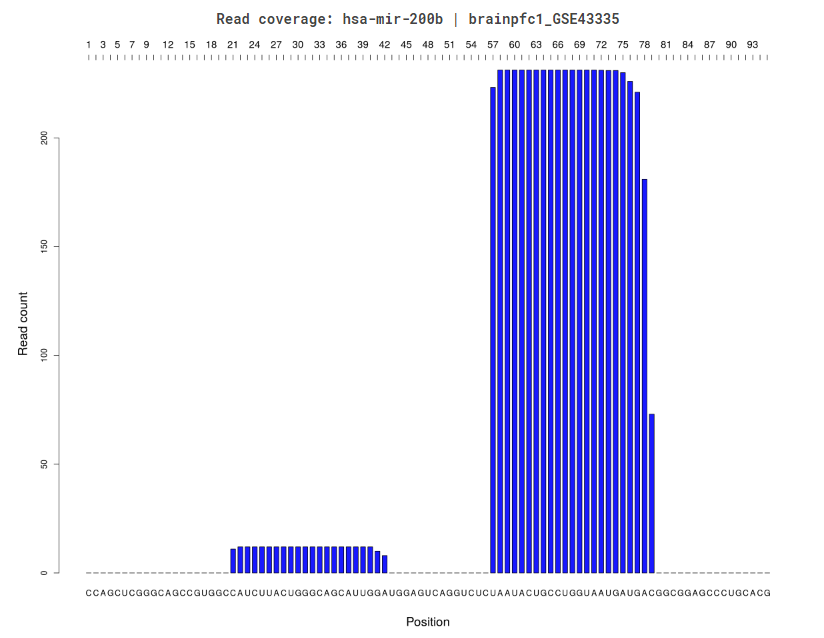
The read coverage plot available for a single tissue or the entire set of tissues, displays the number of raw reads from the chosen tissue along the genomic coordinates of the locus. The sequence is displayed underneath the plot.
Read Coverage Per TissueRead Coverage Profile Across Tissues
Entropy/specific processing
For each processed sncRNA locus, we also describe the processing specificity for both ends of its mature sncRNA products in each tissue using cleavage specificity scores (calculated using CoRAL). This information is available in the tissue-specific section of the entry page along with the read coverage profile for the sncRNA locus.
The entropy score was computed based on the distributions of the 5p and 3p end positions of all sncRNA reads mapped to a given locus, respectively. This entropy feature was designed to capture the specificity (or degeneracy) of RNA cleaving enzymes specific to the production of different types of sncRNAs. For example, the processing of mature miRNA products from precursor miRNAs tends to produce fragments with a more stable 5p cleavage position (low entropy) and more variable 3p end (higher entropy).
All these plots and expression tables for a locus can be downloaded on the same page.
Browse
Small non-coding RNA expression
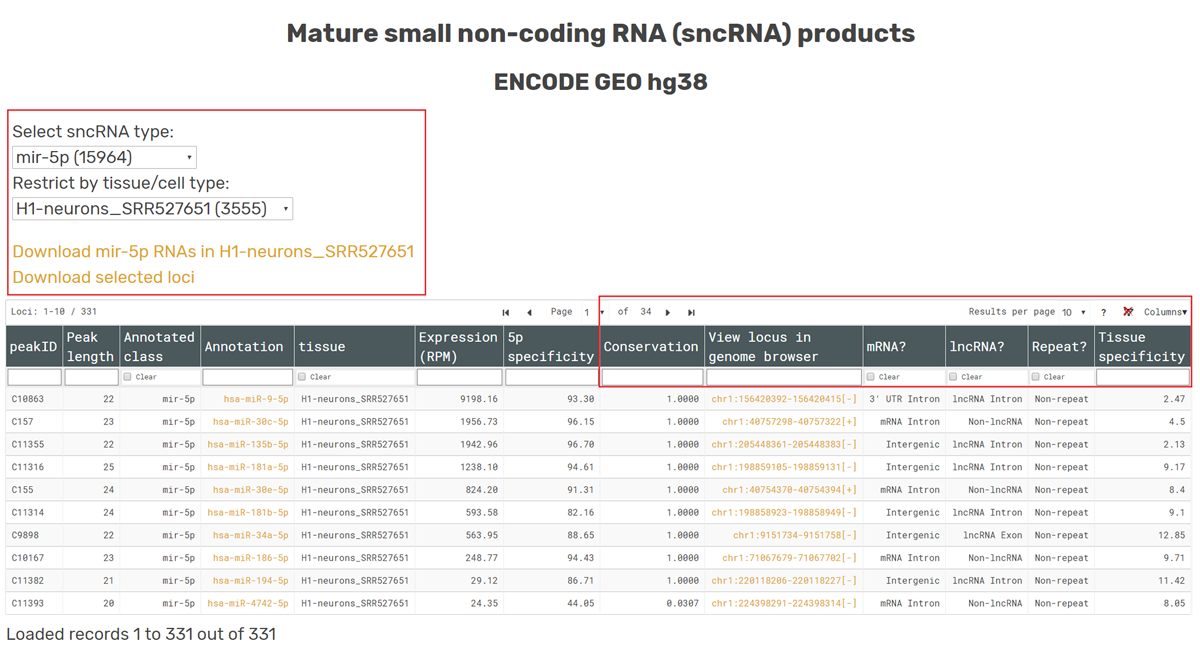
The browsing section provides two tables which allow users to identify either small non-coding RNA genes or mature sncRNA products of interest and download their expression profiles. With the two drop-down menus, users can a)choose which specific sncRNA type they want to look at; and b) choose which tissue/cell type to use for sorting the expression values from largest to smallest. Users can also choose to sort by the average RPM values across all tissues. Note: all genomic coordinates/locations can be sorted.
A snapshot of the small non-coding RNA gene expression table, which contains the expression profiles in RPM for sncRNA genes is shown below:
A snapshot of the table containing sncRNA loci information is shown, with default table view containing expression (6th col), 5’ specificity (7th col), conservation (8th col) and tissue specificity scores (13th/last col). Users can also filter sncRNA loci that colocalize with mRNA (10th col), lncRNA (11th col) or repeat regions (12th col). Additional columns can be added/removed for sorting and filtering (top right corner of the table). Users can download all sncRNA loci for selected RNA class and tissue and/or filtered set of loci (‘Download’ links above the table).
View genomic region
In the text box, users can input a genomic region with chromosome, start and end coordinates, and strand information. The information for this region will be displayed in a new page containing the information described in the "Outputs" section.